An Everyday DNA blog article
By: Sarah Sharman, PhD, Science Writer
Illustrated by: Cathleen Shaw
Genome sequencing technology has revolutionized the study of life and biological processes. The ability of scientists and physicians to analyze the DNA sequences of whole genomes has led to groundbreaking discoveries in wide-reaching fields from drug discovery to cancer research, population genetics to variant studies, agricultural research to infectious disease research.
The first draft of the human reference genome was published by scientists in the Human Genome Project (HGP) twenty years ago this week. The completion of the first human reference genome is often referred to as the beginning of the genomics revolution and is one of the most monumental achievements in biological research. But what is a reference genome and why is it important?
What is a reference genome?
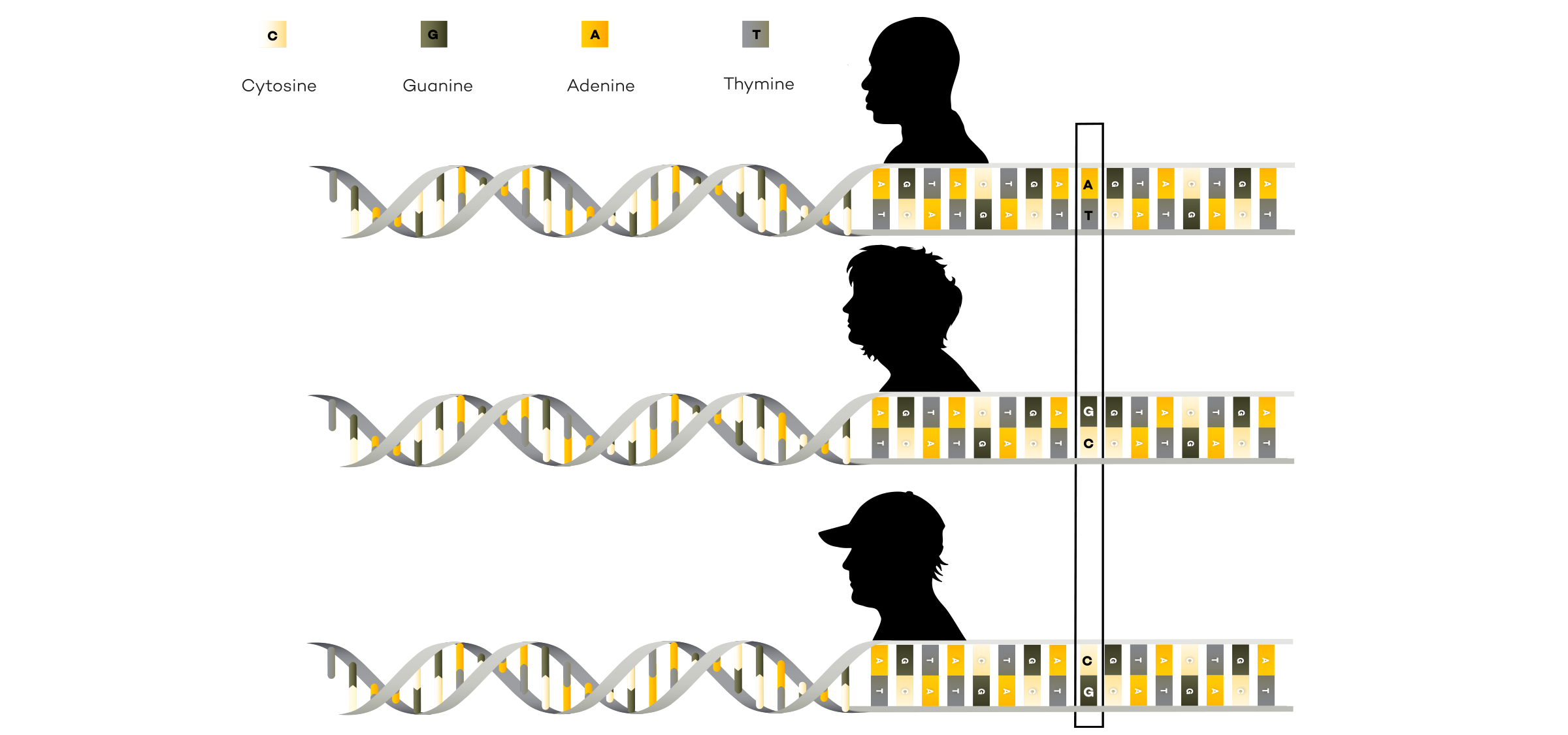
A reference genome is a representative example of a set of chromosomes for a species, ideally produced from the DNA of one member of that species. However, the original human reference genome was derived from the DNA of several volunteers.
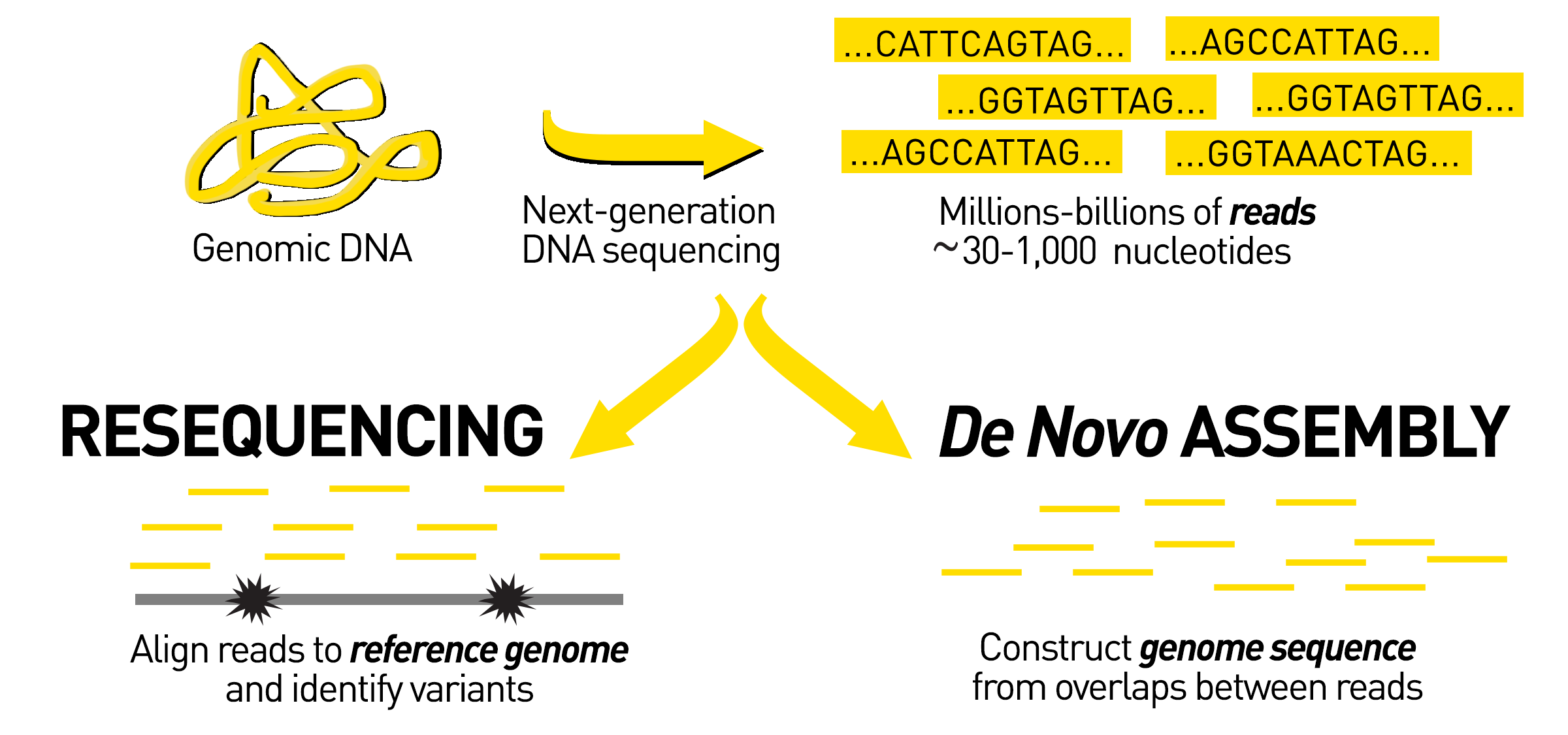
 The availability of reference genomes is important in the field of genomics because they serve as a template for scientists when they are sequencing other members of the same species. Sequencing an individual from a species that already has a reference genome is called resequencing. By using the reference genome as a guide, the scientists can more easily determine where individual pieces of the newly sequenced genome belong when they are assembling the sequence.
The availability of reference genomes is important in the field of genomics because they serve as a template for scientists when they are sequencing other members of the same species. Sequencing an individual from a species that already has a reference genome is called resequencing. By using the reference genome as a guide, the scientists can more easily determine where individual pieces of the newly sequenced genome belong when they are assembling the sequence.
Reference genomes also serve as a point of comparison when looking for disease or trait causing variants. Scientists can compare the DNA of an organism to the reference genome to pinpoint changes in the DNA sequence and do further studies to determine if the variant is responsible for disease or other traits.
While reference genomes are very useful to the genomics field, it is important to keep in mind that reference genomes are just an idea of what a normal organism’s genome should look like. For example, about 70% of the sequence of the original human reference genome produced by the public HGP came from a single anonymous male donor from Buffalo, New York, which cannot possibly represent the DNA of all mankind. Scientists are trying to remedy this lack of diversity through projects like All of Us, an initiative that is inviting one million people across the U.S. to submit health data, including a DNA sample, to help build a diverse health and genomic database.
For plant genomes, scientists are moving away from having only one reference genome for a species and instead are embracing the pan-genome, a collection of all the DNA sequences that occur in a species. Pan-genomes present DNA sequences that are shared between members of a species, but also DNA sequences that are unique to individual members. We will discuss pan-genomes more in depth in an upcoming blog post.
How do we create a reference genome?
So how do we create a reference genome without an existing genome to use as a template? Reference genomes are prepared through a process called de novo genome sequencing. De novo sequencing occurs when scientists sequence and assemble a genome from scratch without using a reference genome for alignment.
To begin de novo sequencing, scientists create many copies of the DNA of interest and chop up, or fragment, the large genome into smaller pieces. The smaller pieces are sequenced using short-read sequencing, long-read sequencing, or a combination of both techniques. Then all of the sequenced pieces, called reads, are assembled back together to produce the whole genome sequence.
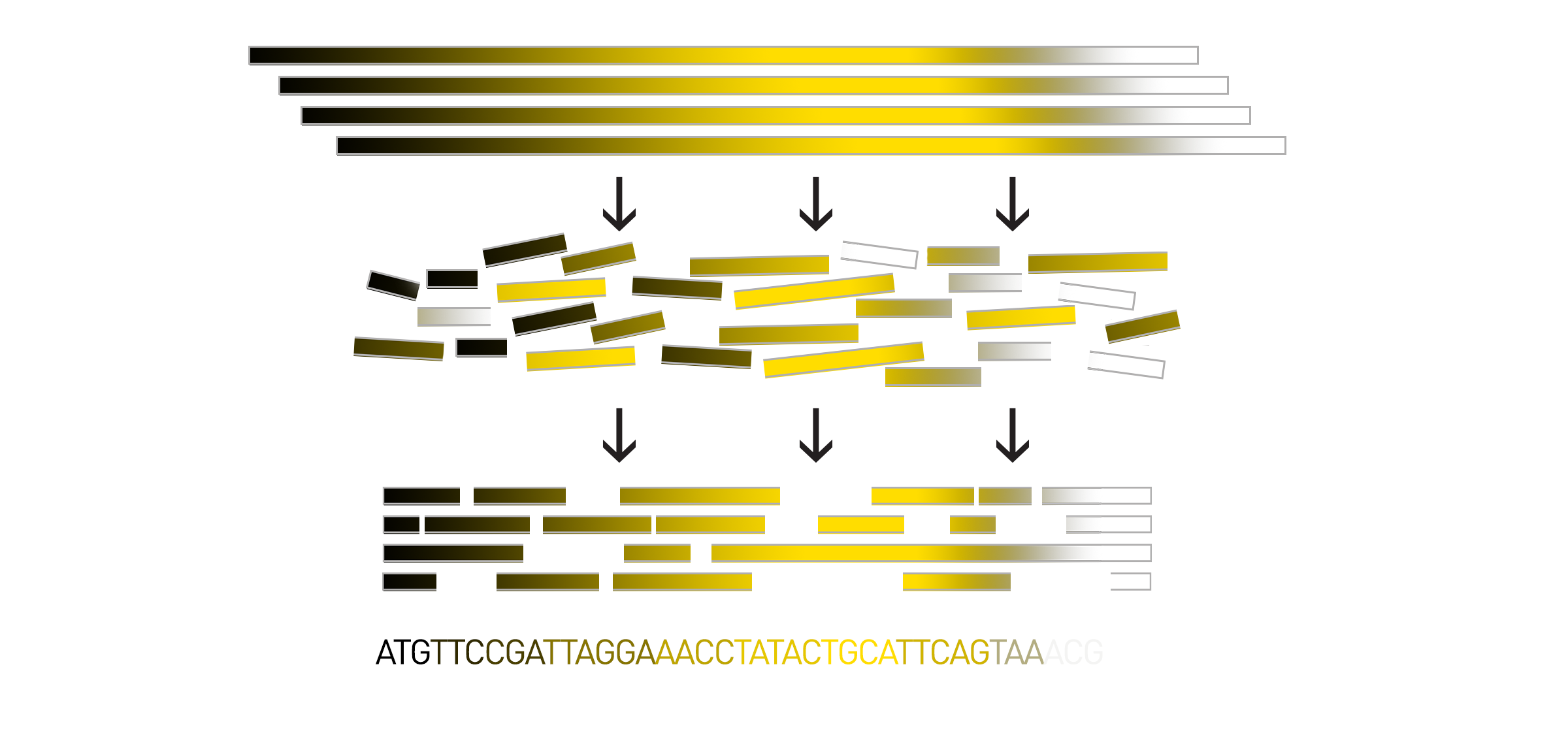 As I previously mentioned, in de novo assembly there is no reference template to help guide the reassembly of the reads. Instead, scientists rely on computational programs called assemblers to help them reassemble the pieces of sequenced DNA. The order of the reads can be computationally inferred by detecting overlapping regions within the reads. The more similarity between the end of one read and the beginning of another, the more likely they are to have originated from overlapping sections of the genome.
As I previously mentioned, in de novo assembly there is no reference template to help guide the reassembly of the reads. Instead, scientists rely on computational programs called assemblers to help them reassemble the pieces of sequenced DNA. The order of the reads can be computationally inferred by detecting overlapping regions within the reads. The more similarity between the end of one read and the beginning of another, the more likely they are to have originated from overlapping sections of the genome.
The computer program pieces short reads together into larger, overlapping chunks which are called contigs, short for contiguous sequence. Then all of the potential contigs are compared by the computer program and the most frequently occurring base at each location of the genome is chosen as the consensus base for that location. In another approach, the computer program runs an algorithm that finds the most likely connection of the contigs which it assembles into larger pieces called scaffolds. Sometimes, there is enough genomic information for the assembler to go one step further and connect scaffolds into complete chromosomes.
 To achieve the most accurate, high-quality genome assembly, it is important to build a gapless assembly. This means that there are no missing pieces in the genome. Gapless assembly can be achieved in a number of different ways such as using longer reads to produce longer contigs, or using a combination of deep sequenced short reads and long reads. Besides gapless assembly, other indicators of the quality of a genome include overall reassembled sequence size compared to the estimated genome size and sequence contiguity (the number of contigs and contig length).
To achieve the most accurate, high-quality genome assembly, it is important to build a gapless assembly. This means that there are no missing pieces in the genome. Gapless assembly can be achieved in a number of different ways such as using longer reads to produce longer contigs, or using a combination of deep sequenced short reads and long reads. Besides gapless assembly, other indicators of the quality of a genome include overall reassembled sequence size compared to the estimated genome size and sequence contiguity (the number of contigs and contig length).
How are scientists using de novo sequencing and reference genomes?
Scientists at the HudsonAlpha Institute for Biotechnology have a long and fruitful history of sequencing complex genomes, dating back to the Human Genome Project. Using the ever-advancing next-generation and third-generation sequencing technologies, the HudsonAlpha Genome Sequencing Center (GSC), led by HudsonAlpha Faculty Investigators Jane Grimwood, PhD and Jeremy Schmutz, are masters at generating complex plant genomes. As of early 2021, they have sequenced reference genomes for more than 175 plants—approximately half of the plants sequenced as high-quality references worldwide. The reference genomes produced by the GSC have helped researchers find beneficial traits that were lost in domesticated crops, trace the evolutionary history of crops, identify genes related to desirable traits, and study environmental effects on domestication.
For example, in one study the researchers compared the genomes of wild and domesticated cotton species to try to understand more about cotton fiber development. During the study, the GSC team sequenced and assembled reference-grade genomes for the two major lineages of domesticated cotton and compared them with three wild cotton genomes. By comparing the sequences, they identified unique genes related to fiber and seed traits in two domesticated species, and found that wild cotton has more disease resistance triggers than domesticated cotton varieties.
Through the assembly of a high-quality reference genome for the grass Setaria viridis, the GSC team and colleagues identified a gene related to seed dispersal in wild plant populations for the first time. They identified a gene called Less Shattering 1 (SvLes1) which is involved in a process called shattering, or seed dispersal. Shattering is critical for plants in the wild to create offspring, but it is an undesirable trait for domesticated crops because it leads to reduced harvest yields. This newly discovered shattering gene variant could be targeted as a mechanism to turn off shattering in other plants, thus improving their yield.
More recently, the team completed a high-quality reference genome for a plant useful for producing biofuel called switchgrass that has a very complex genome. The reference genome took the team nearly ten years to complete because they kept “upgrading” the genome sequence using new sequencing technologies as they emerged. The newest version of the switchgrass genome allows scientists to spot regions in the genome that are associated with important traits like cold tolerance. Once these regions are identified, breeders can use them to develop new strains of switchgrass.

