An Everyday DNA blog article
Written by: Sarah Sharman, PhD, Science writer
Illustrated by: Cathleen Shaw
The first complete genome of a free-living organism, the bacterium Haemophilus influenzae, was completed in 1995, quickly followed by the genomes of many other model organisms, including the yeast Saccharomyces cerevisiae, the roundworm Caenorhabditis elegans, and the plant Arabidopsis thaliana.
By the time the first near-complete human genome was published in 2003, it seemed that scientists had the tools to sequence the whole tree of life. Creating one complete genome for every species would give scientists the key to unlock the inner workings of all of life on earth, right? It turns out, it is not that simple.
Over the past decade, large-scale sequencing projects have revealed high levels of genomic changes across the tree of life, suggesting that single genomes do not accurately represent the full genetic diversity of a species. Let’s learn how scientists are working to increase diversity in genomic sequencing.
What is a pangenome?
In the early days of genetics and genomics research, DNA sequencing was very expensive and time-consuming. As such, scientists created single reference genomes—generated using one individual from a species—to represent the entire species. Reference genomes are used as a point of comparison when identifying disease- or trait-causing DNA changes, comparing sequence similarity in genes across species, and many other aspects of genomic research.
With advances in sequencing technology, scientists now have the ability to sequence more individuals within a species faster and more affordably. Using the growing number of unique genomes, scientists have begun constructing collections of genomes, called pangenomes, to help represent the full genetic diversity of a species.
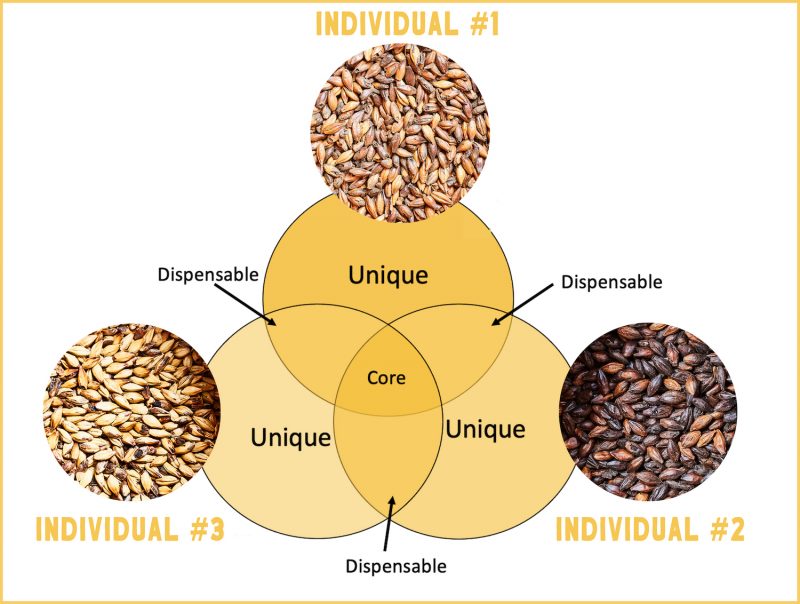
Pangenomes present all of the genes and DNA sequences within a species. They are created by sequencing many members of a species and comparing their genomes. Pangenomes show the complete genome information of a species as well as the existing genetic diversity that is characteristic of the species.
Within a pangenome, the core genome includes the genes and genetic changes that are shared by all of the individuals in a species. The dispensable genome is the portion of the genome that is only in a subset of individuals or is unique to one individual. This contains, for example, genes that have been selected by adapting to certain environmental conditions.
What can pangenomes teach scientists?
Pangenomes were originally constructed for species of bacteria and have proven useful in the field of microbiology. For example, a detailed knowledge of microbial diversity can help make better vaccines. A deeper understanding of the core genome shared by all influenza viruses could help create a universal flu vaccine, increasing the efficacy of the flu vaccine and potentially eliminating the need to be vaccinated each year.
Genetic variation is also linked to growing numbers of genetic disorders. This makes a strong argument for a view of the human species that takes genetic changes into account. The Human Pangenome Project is working to develop a better representation of sequence diversity in the human population, starting with creating 350 diverse human genomes.
The use of pangenomes is now gaining traction in other fields, particularly in the field of plant biology where plants often genetically adapt to environmental conditions. In crop plants, differences in genes have implications on disease resistance, metabolite production, stress response, and other biological phenomena.
At the HudsonAlpha Institute for Biotechnology, scientists in the Center for Plant Science and Sustainable Agriculture are embracing the importance and usefulness of pangenomes in their research. By looking at many genomes for a species, the scientists have recently made discoveries in plants such as switchgrass, green millet, and barley that would not have been possible without the pangenome. Let’s dive a little deeper into the barley pangenome.
Case study: barley pangenome
 In the United States today, barley is predominantly used for malting and animal feed, although certain types of barley are also part of the human diet. However, in developing countries barley is still an important part of the human diet because of its high nutritional value and wide adaptability to a diversity of climates and environments.
In the United States today, barley is predominantly used for malting and animal feed, although certain types of barley are also part of the human diet. However, in developing countries barley is still an important part of the human diet because of its high nutritional value and wide adaptability to a diversity of climates and environments.
It is predicted that by the year 2050 the world’s population will be 9.7 billion people, nearly 2 billion more than currently inhabit our planet. The world’s land and water resources are finite with population growth placing pressure on these valuable resources.
In order to meet the increased need for food without depleting land and water resources, we need crops to produce more output on existing land with fewer inputs like water and fertilizers. So how do we increase the yield potential of crops like barley without taking up more land mass? The answer might lie within the plants’ genomes.
Understanding the DNA variants that barley needs to produce useful traits, such as increased yield and ability to survive extreme weather, can help crop breeders create optimized varieties of barley suitable for growth on marginal lands with little resource input and maximum seed output.
The barley genome is huge, clocking in at nearly 5 billion base pairs (for reference the human genome is about 3 billion base pairs). Although the first barley reference genome was published in 2012, it is only representative of one barley variety. There is still much work left to be done to unravel the barley genome at a deep enough level that breeders can confidently use the genetic information to make breeding decisions.
Researchers at the HudsonAlpha Genome Sequencing Center recently contributed their genome sequencing and analysis expertise to a study that brings them one step closer to understanding the entire barley genome. In the study, the researchers created a barley pan-genome by sequencing many varieties of barley.
The team surveyed approximately 22,000 barley seeds in a gene bank and chose 20 varieties for genome sequencing to serve as representatives of global barley diversity. By analyzing the 20 newly sequenced barley genomes, the group observed that two barley varieties can differ in the number of genes and in the arrangement and orientation of large parts of individual chromosomes, called structural variants.
These types of variants make breeding new combinations of desired traits extremely difficult. By knowing the locations of structural variants, breeders can choose the appropriate barley varieties to cross to achieve desired traits in the offspring.
Although the team still needs to sequence several hundred more barley varieties, including wild barley, this pangenome brings them one step closer to understanding the complete picture of the barley genome and its diversity.

